Маленькие бактерии и лбы, на которых они обитают
Всем привет!
Так уж получилось, что сейчас немного осваиваю биоинформатические методы исследования.
Попрошу знающих отписаться в комментариях с советами, так как все делал по статьям и протоколам с интернета.
В настоящее время изучаем в основном микробиом почвы и ризосферы, и недавно подготавливал новую 16S библиотеку.
Для подготовки 16S библиотеки нужно :
1. Выделить ДНК
2. Амплифицировать нужный участок ДНК
3. Прикрепить баркоды - специальные метки, разные для ДНК из каждого образца.
4. Секвенирование - считывание последовательности нуклеотидов из ДНК в 16S библиотеке.
Прежде всего необходимо выделить ДНК из образцов (у меня - почва, но это может быть кровь, растительные или животные ткани, кал, на что фантазии хватит). Можно использовать коммерческие киты, или наборы, например, Power Soil для почвы, Blood & Tissue для крови и тканей, от разных производителей. Я использовал Power Soil и все образцы у меня были из почвы, однако захотелось мне проверить, на будущее, как поведёт себя набор для экстрагирования ДНК из почвы со swab образом (типа ватной палочки, смоченной в одном из буфферов для ДНК или ультрачистой воде, которой можно забрать мелкие объекты с ДНК с какой либо поверхности)

Так что я взял такую палочку, смочил в TE буффере и провел себе по лбу. А так как мне интересно узнать эффективность данного набора при работе с не предусмотренным разработчиком источником, я едва касался палочкой кожи.
Выделение ДНК подробно описывать не буду, так как слишком долго, и я это уже описывал ранее в одном из постов со старого аккаунта, который я удалил по личным обстоятельствам. Может, кто меня даже и вспомнит)).
Работал под ламинаром для уменьшения вероятности контаминации образцов (попадания ДНК из других источников).

В одну библиотеку можно включить сразу большое количество образцов. Обычно работают с плашками на 96 лунок, фото с нета для примера.

По итогу имеем плашку, в которой в каждой лунке будет находиться раствор с ДНК одного из образцов. Главное - вести запись, где какой образец! Потом будет поздно. В одной из такой лунок находился и образец с моей кожи на лбу.
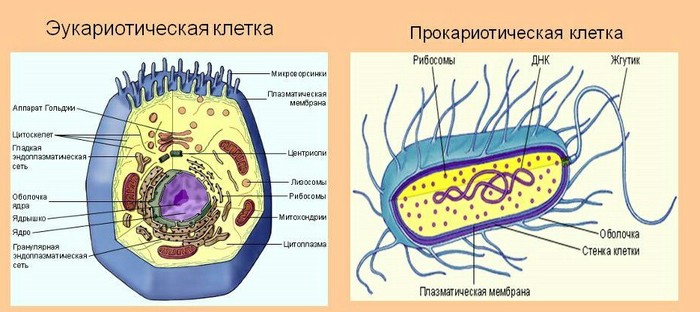
Далее необходимо амплифицировать (умножить количество) целевую ДНК. Так как я готовлю 16S библиотеку, биологи уже поняли, что интересует меня ДНК бактериальных рибосом.
На картинке снизу показано строение бактериальной рибосомы (верхняя) и эукариотической (снизу). Как вы видите, у бактерий малая субъединица рибосомы включает в себя 16S РНК, а мне нужно умножить количество ДНК из извлеченной, ее кодирующую.
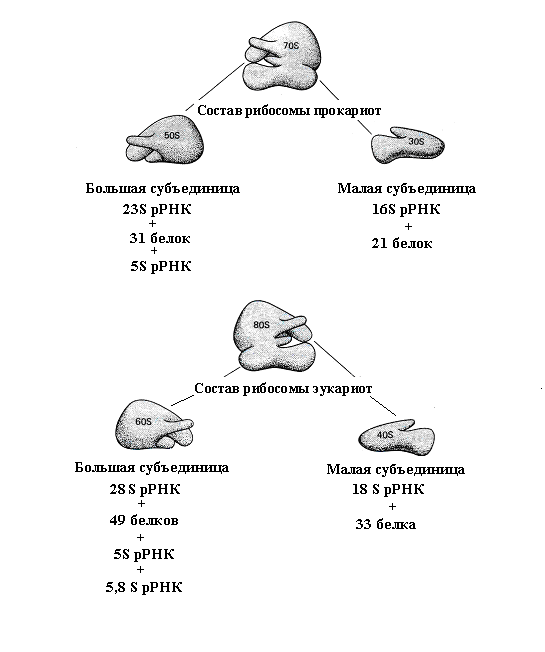
Почему именно этот ген? Именно по нему очень удобно идентифицировать различные бактерии, так как:
- Есть у всех бактерий
- Достаточно консервативен
- Достаточно варибелен для классификации.
Данный ген состоит из консервативных (одинаковых у всех бактерий) и вариабильных участков, по которым и можно различать бактерии.
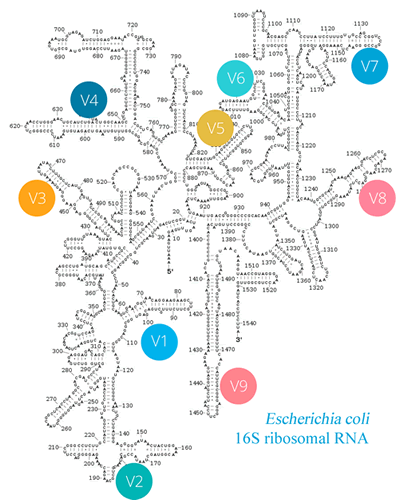
Обычно используют участки V3-V4 или V4.
Я использовал V4. Для этого можно заказать готовые праймеры - фрагменты РНК/ДНК, способные связываться с целевой ДНК, и необходимые для амплификации ДНК.
Амплификация ДНК осуществляется при помощи ПЦР - полимеразной цепной реакции. Ссылка для ЛЛ: https://ru.m.wikipedia.org/wiki/%D0%9F%D0%BE%D0%BB%D0%B8%D0%...
Если кратко, то это реакция, которая позволяет строить новые молекулы ДНК на основе ДНК из ваших образцов. Молекулы ДНК состоят из двух цепочек, комплементарных друг другу, и строятся из последовательности 4-х нуклеотидов - аденина, гуанина, цитозина и тимина. Комплементарность - это способность нуклеотидов из разных цепочек образовывать связь друг с другом - аденин связывается с тимином, цитозин - с гуанином, при помощи водородных связей (нуклеотиды внутри цепочки соединены более сильной ковалентной связью). В итоге в одной молекуле ДНК содержатся две цепочки, которые, хоть и содержат разные последовательности нуклеотидов, комплементарны друг другу и на основе одной из них всегда можно построить другую.
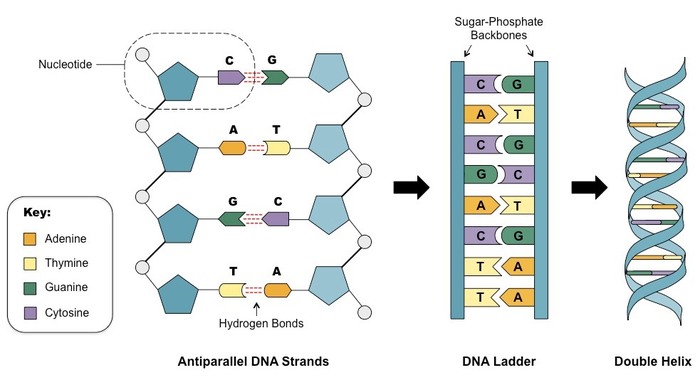
При этом цепочки антипараллельны друг другу - представьте это как два поезда на станции, которые должны отправиться в разные стороны и пока стоят рядом на путях. То есть начало одной цепочки находится рядом с концом другой и наоборот. Это важно, так как строение новой цепочки на основе старой может идти только в одном направлении.
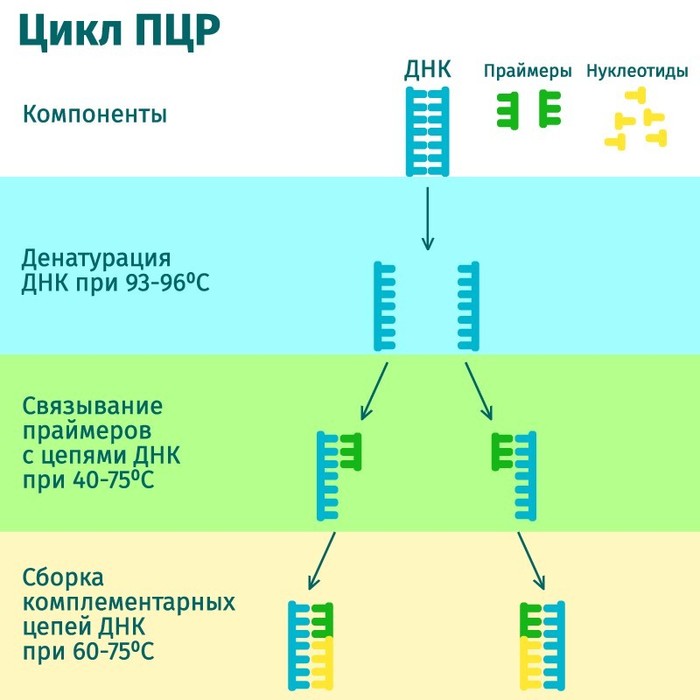
Для ПЦР готовят смесь из ДНК, праймеров, полимеразы (фермент, который строит ДНК), и нуклеотидов. Реакция проводится в амплификаторе, или ПЦР-машине, способной периодически нагревать и остужать смесь.
Так вот, при ПЦР молекулы ДНК нагреваются примерно до 95 градусов, при этом относительно слабые водородные связи между двумя цепочками разрушаются, и они расходятся. Тут нам и пригодятся праймеры - при остывании смеси ДНК может восстановить водородные связи, и праймеры заранее подбираются таким образом, чтобы связаться с нужным нам фрагментов ДНК по принципу комплементарности, затем полимеразы достраивают из нуклеотидов новые цепочки ДНК на основе старых. И так 25-35 циклов подряд.
На выходе имеем опять таки плашку с разными образцами в каждой лунке, но теперь ДНК в основном состоит из целевых фрагментов, в моем случае - V4 из РНК бактериальной малой субъединицы рибосомы.
После ПЦР проводится прикрепление меток - баркодов, в каждой лунке - свой баркод. Получается, что-то типа такой таблицы
Образец 1 - баркод 1
Образец 2 - баркод 2.
Каждый баркод имеет свою уникальную последовательность ДНК, которая при помощи ещё одной ПЦР реакции прикрепляется к амплифицированным при первой ПЦР молекулам ДНК.
После того, как баркоды добавлены, можно объединить все образцы в одну пробирку и секвенировать - прочитать последовательность нуклеотидов в ДНК. Я работал на ISeq. Фото с нета.

После секвенирования я могу скачать архив архивов, каждый из которых содержит имя одного из моих образцов. Для этого и нужны были баркоды - машина считала ДНК из общей кучи, а потом разделила по архивам согласно баркодам.
Данные архивы я обрабатывал программой Qiime2, специально разработанной для изучения микробиомов. Эта программа, или, вернее, среда - настоящий комбайн, с помощью которого можно посчитать статистику, корреляции, определить таксономию, и так далее. И все - бесплатно.
Здесь приведу очень краткий пример, только для одного образца, поэтому 95% функционала Qiime2 здесь показано не будет, только пример таксономической классификации бактерий.
Машина с Ubuntu 18.04 и Qiime2-2019.04 на борту.
Активирую среду в терминале и запускаю Jupyter lab (команды можно запускать и с терминала, но лаб намного удобней)
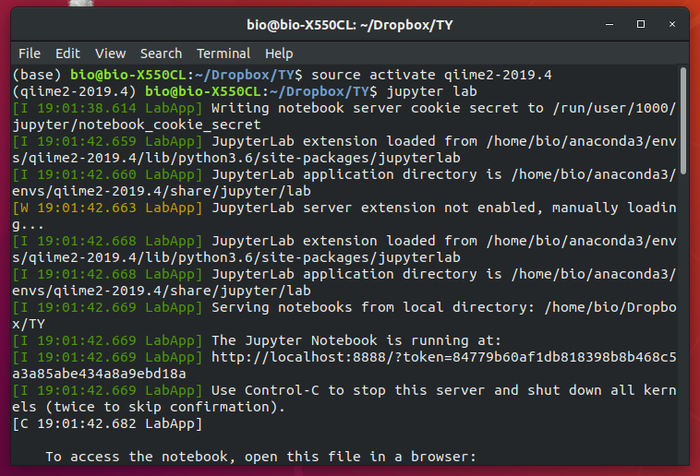
В браузере открывается такое окно. На скрине я уже набрал код для импортирования моих данных в программу. У программы есть сайт, который легко гуглится, с мануалами, туториалами и активным форумом, где вам всегда помогут.
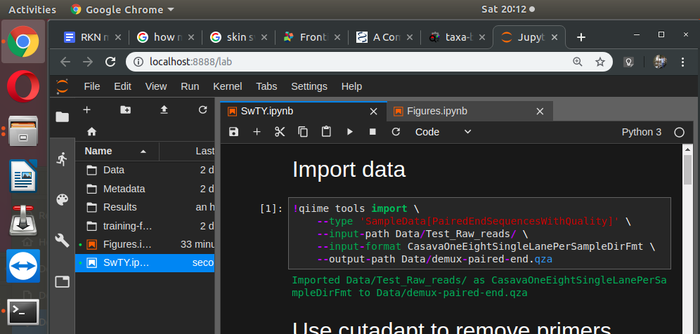
После этого ввожу команды для удаления праймеров из последовательностей ДНК
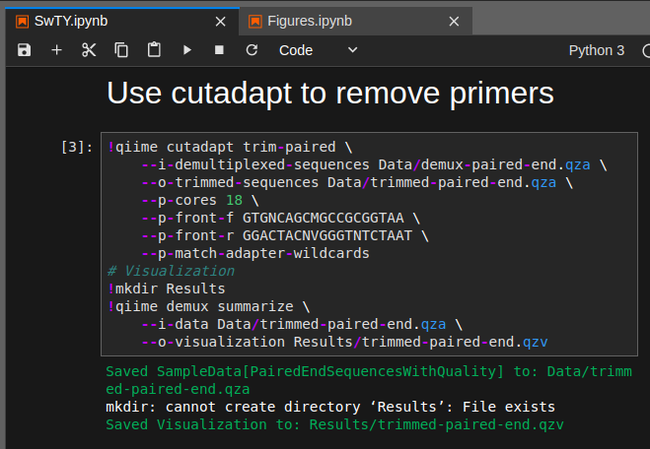
Если нужно, последовательности, считанные с противоположных концов одного фрагмента одной ДНК, объединяются в одну последовательность и фильтруются
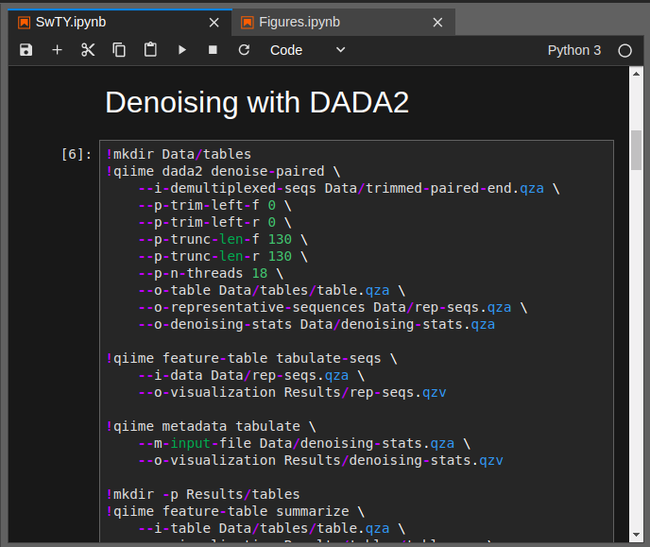
Теперь можно и классифицировать бактерии. Для этого нужна датабаза известных 16S ДНК бактерий - я скачал с сайта Silva последнюю и натренировал на ней ранее классифайер (по инструкциям с сациа), который и использовал на своём образце
В итоге я знаю, какие бактерии живут у меня на лбу
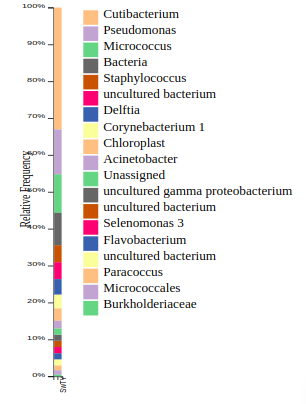
Да, там есть хлоропласты, и я подозреваю, что частично я - растение. Ну или просто слишком много возился с растениями в тот день=).
PS - малое количество бактерий, так как я едва провел палочкой по лбу, и моя задача была проверить, смогу ли я использовать этот набор для почвы для обработки swab образцов - в принципе, можно. В образцах из почвы я получил данные по сотням бактерий с той же библиотеки, и с ними сейчас работаю в Qiime2 над статистикой, альфа и бета разнообразием и пр.
И да, я пропустил этапы очистки ДНК после пцр и нормализацию ДНК, так как слишком много бы получилось.
Кто дочитал - я вами восхищаюсь.