

Синдром Крузона
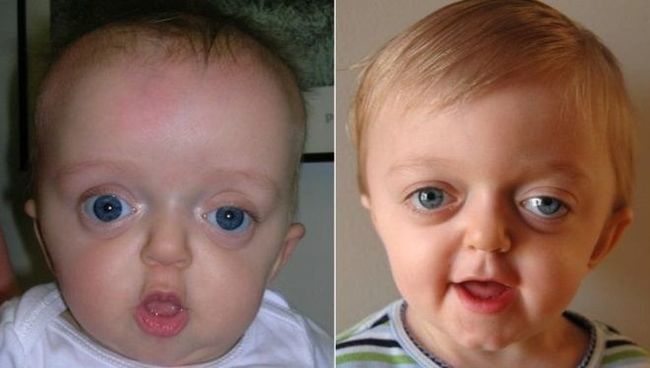
Синдром Крузона — краниостеноз, обусловленный сочетанием недоразвития костей черепа и преждевременным зарастанием черепных швов, что проявляется изменением формы мозгового и лицевого черепа. Развитие деформаций лицевого скелета при данном заболевании обусловливает развитие экзофтальма, роговично-конъюнктивального ксероза и может привести к спонтанному вывиху глазных яблок из орбит.

Частота заболевания оценивается как 16,5: 1 000 000 новорожденных.
Выделяют два варианта синдрома Крузона: классический и с черным акантозом. Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме — он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов (более 35 мутаций), что снижает его стабильность. В основном поражает элементы скелета. Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка.
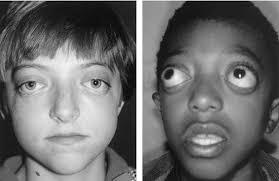
Причины синдрома Крузона с черным акантозом несколько иные — он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями — Ala391Glu. Это тоже миссенс-мутация, изменяющая структуру белка-рецептора.
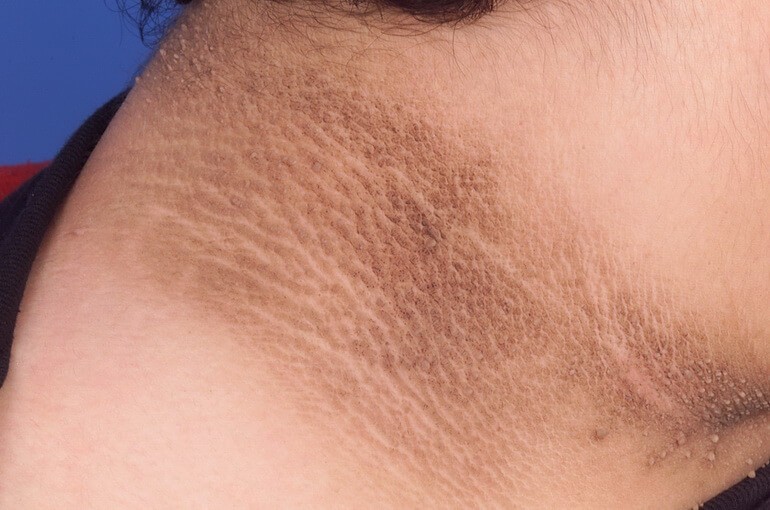
Синдром Крузона с черным акантозом характеризуется аналогичными изменениями со стороны лица и черепа. При этом некоторые исследователи отмечают, что данная форма заболевания протекает в целом тяжелее и характеризуется повышенной частотой осложнений.
Клинические проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни.
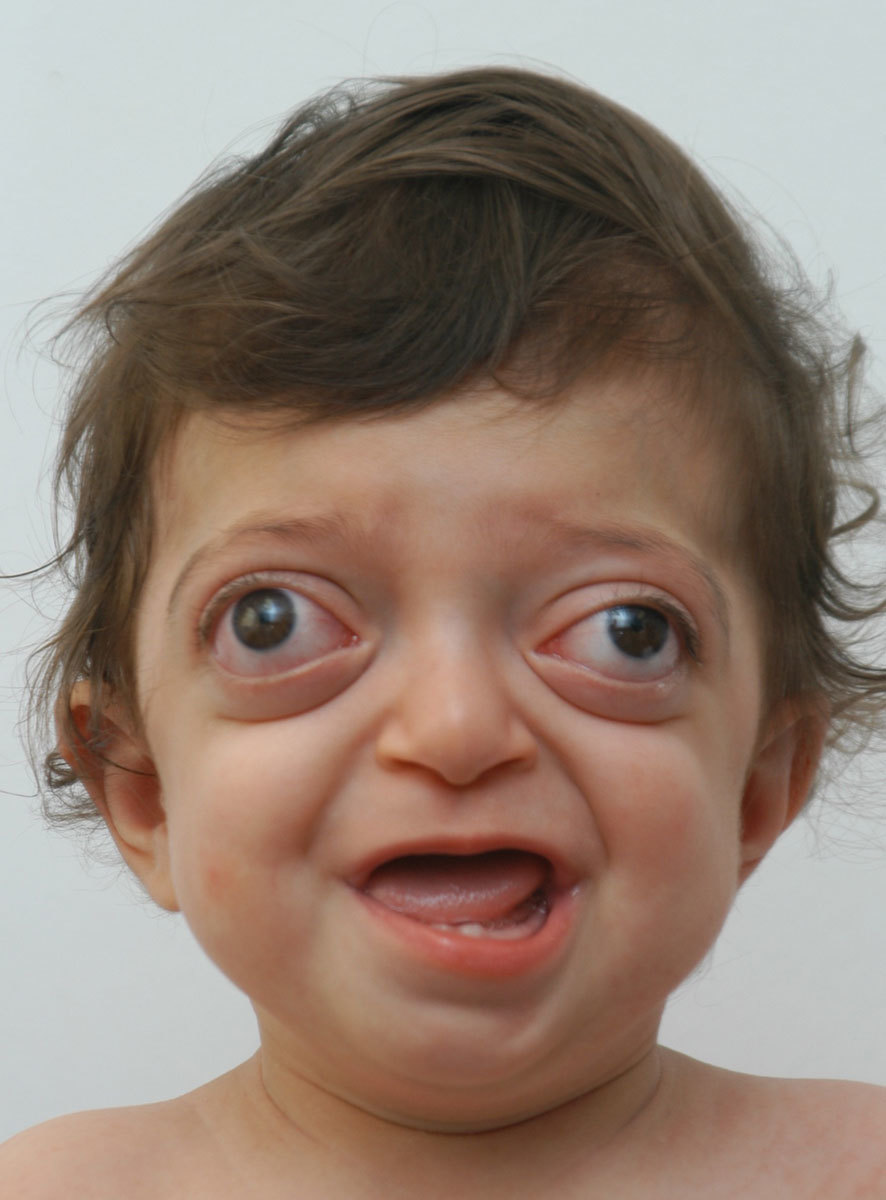
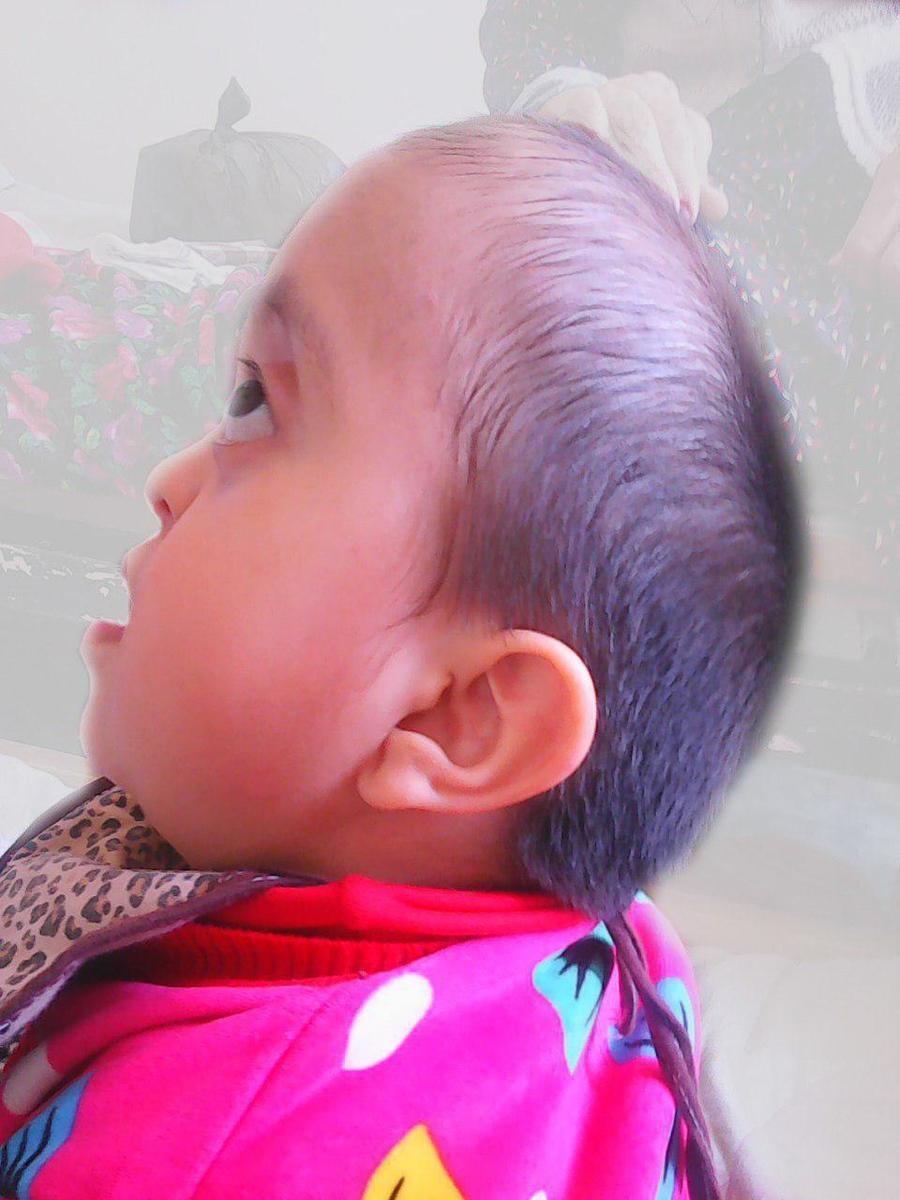
Ниже приведен классический вариант синдрома Крузона. В неврологическое отделение поступила девочка 2013 года рождения с жалобами матери на необычный вид ребенка: пучеглазие, «некрасивую» форму головы, задержку развития, судороги.

Из анамнеза: ребенок от первой беременности, первых родов, родилась в срок с весом 4200г с оценкой по шкале Апгар 4 балла. Беременность протекала на фоне резко выраженного токсикоза в первой половине беременности, угрозы на втором месяце беременности. Мама перенесла грипп в первом и третьем триместрах беременности. Лечение не получала. Брак не родственный. Родители молодые, внешне здоровые. Родственные браки отмечены по линии отца ребенка. Наследственные заболевания отрицают. Родословную составить не удалось, родители в разводе. Состоит на учете у невропатолога с 3-х месячного возраста по поводу гидроцефального синдрома. В 1,5 года выставлен диагноз: Синдром Крузона.
Отмечена задержка физического, нервно-психического и психомоторного развития ребенка. Начала сидеть в течение последних трех месяцев. Ходит с поддержкой. Речь отсутствует, но начала издавать звуки. Не понимает обращенную к ней речь. Пытается следить за движущимися предметами. Периодически нистагм и расходящееся косоглазие. Выраженный экзофтальм. Веки не смыкаются. Не моргает. Со слов матери, когда «глаза устают» и во время сна девочка принимает колено-локтевое положение, уткнувшись лицом в подушку. На осмотр и звуки реагирует вяло. Не дифференцирует где свои, где чужие, но мать чувствует. Зрачки D=S, округлой формы. Движения глазных яблок безболезненное. Лицо симметричное. Язык по средней линии. Глотание не нарушено. Парезов и параличей конечностей нет. Менингеальных знаков и патологических рефлексов нет.
Уровень медицины нашего времени позволяет своевременно диагностировать генетическую патологию. Для этого необходимо усилить санитарно-просветительную работу среди населения о последствиях родственных браков, путем бесед, показа видеороликов, а также обязательный 100% скрининг перед заключением брака, и беременным женщинам для предупреждения и раннего выявления врожденной и наследственной патологии.